
❝ I am Wafic Zein, 22 years old, from Lebanon. I was diagnosed with Duchenne Muscular dystrophy at 4 years old. Duchenne Muscular Dystrophy (DMD) is a rare progressive disorder.
Muscles become weaker over time until it affects the whole body. Approximately one in every 5.000 boys is born with the disease. It is caused by a mutation on the X-chromosome. this is why only a few girls are affected.
I was hardly following my friends and my younger sister during play time, falling frequently very slow on climbing stairs and I was never able to jump. However, I was very active at home and at school. My diagnosis was after several tests, doctors' visits, genetic test and biopsy. I didn’t know much at this young age, but my parents told me by the time that my muscles are weak, and the therapies that I undergo are essential to preserve them and to improve my abilities.
My parents had never heard of the disease before and sadly knew nothing about it. First, they found themselves alone and looking for information that can enlighten the reality of the diagnosis Whereas, I found myself unique as a little boy. It was not long after my parents decided to make changes in our lives and all the other little boys and girls who were robbed of a natural healthy life!
Land for hope was founded, a support group that spread awareness, helps, and supports patients and families affected by neuromuscular diseases. During the family gathering I had the opportunity to meet many boys affected by DMD just like me and to make a lifelong friendship and support. I am not alone nor unique anymore! Sadly, at every stage of my disease progression, I lose my abilities and the challenges increase. But I am lucky to have my family by my side to support me and increase my strength to keep fighting and preserve my smile and hope!
❞

❝ The 30th of Jan 2020 was the day that changed our life forever. As we'd been blessed by having our little miracle Aram.
Our story started when Aram was 18 months after he started walking, we noticed his both knees were bending backward, a medical condition known as ((genu recurvatum)), at first it was mild, but by time this deformity began to be more obvious as Aram got older. As parents and physicians, we knew that there was something wrong with Aram's gait.
The long story short, we consulted an Iraqi pediatric orthopedist, who examined & checked Aram limbs and joints. The orthopedist explained to us that Aram has got a phenomenon named "generalised hyperlaxity", which is familial normal variation in individual ligaments, but what made him more concerned was the tightness that he noticed in both Aram's ankles. This orthopedist was the first person who suspected that Aram may complain of a neurological problem.
Aram had been referred to a Pediatric neurologist to exclude any possibility of Spastic Foot, which is a hint to a brain injury such as in cerebral palsy. We consulted the neurologist and during examination the neurologist at that time excluded spastic Foot and excluded any neurological abnormality, and Aram left to be followed up after 2 months.
Unfortunately, Aram condition didn’t get any better during the following 2 months. Living in Iraq with limited medical facilities is a challenge, add to that a rare case as Aram condition. After taking many unsatisfactory medical consultations, and doing various investigations such as serum CPK, vitamin D and Serum Ca levels, X-ray and EMG which all turned out to be negative, we had a raised suspicion that it could be a rare genetic disorder.
So we decided to get rid of this minor possibility by doing the genetic tests. Therefore, we travelled to Turkey to meet with one of the most recommended cytogenetic specialists, who welcomed us and wondered why we ask for a genetic testing as Aram was looking well and there was no reason to suspect genetic disorder. However molecular Karyotyping, and whole exome genetic study were sent abroad to Germany/CENTOGEN LABS, all the investigations were negative for the presence of a definite disease. To this moment I still hearing the voice of the Turkish cytogenetic specialist on the phone congratulating me “your baby boy is normal, your boy is healthy, nothing wrong with him” she said.
During (2021-2022) Aram kept on follow up as normal child, but he didn't improve, he had an imbalance issue (which was one of earlier signs of his actual disease), he avoided walking for long steps scared to fall and getting hurt. Later, Aram wore a Complete brace KAFO for 24 hr/day for 3 months to stop increasing the genu recurvatum, but he didn't find any benefit rather than stopping deformity increments. We even tried 3 months intensive physiotherapy to fix the reduced muscle tone and increased lower limb spasticity.
Anyway, in Ramadan 2022, Aram’s physiotherapist mentioned that when one committed to physiotherapy for 3 months but still couldn’t get the expected results then it’s something progressive and this could be seen in rare genetic disorders. Those two words were the knife that stabbed me in my heart. I was shocked, refused to believe that the genetic test that had been done was simply wrong. During the last 10 months I fought and tried my best not to stand in that position where a specialist could tell me ((sorry, you are late, there is nothing to be done for your son)).
We booked an appointment with a pediatric neurologist that specializes in genetic disorders in Lebanon/ Beirut. Sadly, once the neurologist saw Aram and took the medical history, Aram condition diagnosis was revealed and proved by investigating serum enzymes assay, MRI imaging and repeating the genetic tests in Germany using the same blood samples that had previously taken 10 months ago in Turkey.
After exactly one-year medical journey, Aram was unfortunately diagnosed with a rare genetic disorder involving the white matter in the brain causing demyelination of the cells due to accumulation of sulfatide toxins that destroy the myelin sheath, this disorder called Metachromatic Leukodystrophy (MLD) which is rapidly progressive and carries a poor prognosis, with no treatment for symptomatic patients but supportive one. At first It’s hard to deal with this life changing situation, Aram had to remove his gallbladder due to this disease, also had a gastrostomy tube, despite the fact that he still able to eat orally.
However, with faith we’ll go through this battle, no matter what happens as Aram is our blessed gift. We still hope for a better chance for symptomatic children to get treated.
Our experience recommends that there should be screening tests for newborns, which may provide a better future for affected families. Also, there’s no investigation is precisely accurate. I wish they got the genetic map correct from the first time; it would change the whole story.
I hope my story will inspire all (MLD) patients and all rare diseases community. Pray and support my angel Aram's.
❞

❝ اسمي فرح بودرة، عمري 22 سنة، فنانة تشكيلية من مدينة الحسيمة بالمغرب. أعاني من مرض ضمور العضلات الشوكي وهو مرض نادر يصيب عادة الأطفال ويسبب ضعف العضلات.
عندما كنت في الثانية من عمري، لاحظ والداي سقوطي المتكرر والصعوبات التي أجد في المشي أو الركض مثل باقي الأطفال، فقلقا على صحتي وقررا أخذي للأطباء لفهم ما يحدث. وفي تلك الفترة كان المرض غير معروف بشكل جيد.
بعد رحلة طويلة وشاقة من الفحوصات لدى الكثير من الأطباء، تم أخيرًا تأكيد إصابتي بضمور العضلات الشوكي النوع 3، وأخبر الأطباء عائلتي بأن هذا المرض غير قابل للشفاء. كان خبرا قاسيا على والدي، ومع ذلك قررا عدم الاستسلام ودعمي بكل قوة ومنحي أفضل حياة ممكنة.
عندما بلغت سن التعليم التحقت بالمدرسة، لكن سرعان ما تدهورت حالتي الصحية بسبب المرض وفقدت تماما القدرة على المشي فاضطررت إلى الانقطاع عن الدراسة. رغم هذه الصعاب قررت أن أستمر في التعلم بطريقتي الخاصة، فصارت الروايات والكتب رفاقي الذين أستكشف من خلالهم العالم. خلال هذه الفترة اكتشفت موهبتي وشغفي بفن الرسم وتحديدا فن " البورتريه"، فصرت أقضي معظم يومي في الرسم الذي وجدت فيه وسيلة للتعبير عن مشاعري وأحلامي، أصبح الفن ملاذي ومصدر قوتي للتغلب على صعوبات الحياة ومواجهة المرض.
مع تقدمي في العمر، بدأت صعوبات الحركة تتزايد كما أن المرض سبب لي اعوجاجا في العمود الفقري إضافة إلى متاعب في التنفس، وفي عام 2018 سافرت مع عائلتي لفرنسا من أجل الخضوع لعملية جراحية معقدة لتصحيح العمود الفقري، والحمد لله تمت العملية بالنجاح وتحسنت حالتي نسبيا حيث أصبح بإمكاني الجلوس بكيفية مريحة كما تحسن قدرتي على التنفس.
بعد ذلك وبتشجيع من أسرتي، قررت استئناف تعليمي من المنزل، كان هذا القرار الخيار الأفضل لي. أواصل الدراسة بكل عزيمة وإصرار وأحرص على اجتياز الامتحانات المدرسية نهاية كل سنة، وطموحي هو الحصول على شهادة عليا بالموازاة مع مسيرتي الفنية.
عبر مساري الفني، شاركت في العديد من المعارض الافتراضية وفي سنة 2022 شاركت في معرض واقعي. أدركت أن تجربتي في التعايش مع المرض ورحلتي كفنانة يمكن أن تكون مصدر قوة وإلهام للأشخاص الذين يواجهون تحديات بسبب الإصابة بأمراض نادرة. لذلك أواصل رحلتي الفنية، وأشارك شغفي مع العالم.
الفن أعطاني رسالة وفرصة للتأثير، ورسالتي هي عدم الاستسلام للمصاعب والعقبات التي تواجهنا في الحياة، فمتى تحلينا بالعزيمة والصبر يمكننا أن نصنع المعجزات وأن نحول تجاربنا المريرة إلى لوحات فنية جميلة تحمل بين ألوانها رسائل الأمل والإلهام.
❞
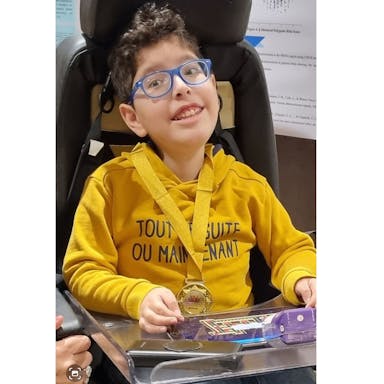
❝ It all started 10 years ago - 29th May 2011.
My mum and dad were happily trying to attract my attention from behind the glass of the newly born babies room - looking back now in their minds that everything was going to change.
My name is Shadi. In Arabic it means, “ A Person with a beautiful voice” ,isn’t it strange that it took me around 4 years to say my first word?! It’s ok for me, since during this period I achieved so many other remarkable milestones - I’ve improved my capability to hold my head up, and I learned how make the eye contact with my parents, and most importantly I proved all the medical doctors telling me I would have live the rest of my life blind, wrong!!
In the first weeks after I was born, my aunt noticed that I was so “floppy” and my eye pupils where not following her. Indeed, my mother noticed this before, but she was not able to believe that I looked “different”, I am not her first baby, and she knew how a baby should react to others.
I had my first MRI experience when I was 5 months old. It was very scary, lonely and loud inside there. My parents waited impatiently, hoping that the MRI result would show something different from what the doctor called: ( Dandy-Walker Syndrome). I am not a doctor, and at that age, I wasn’t able to speak and understand English like now, but my parents' faces and their tears were more eloquence than any language!
It wasn’t easy, but my parents had two options: either to surrender and accept the shocking fact as it is, or to fight with every possible way. They chose to fight, and I said:” count me in, guys!”
Over years, my parents, my brother, my grandmother, along with a great effort by my teachers, speech therapists and occupational therapists, and a lot of gracious prayers from my family and all who knew me, I am able to to balance myself, use my hands, speak English and Arabic, do basic math, and most importantly express myself and show my personality!
In 2016, doctors diagnosed me with MEB disease (Muscles- Eye- Brain) disease, which is an extremely rare genetic disease, caused by mutations in the POMGNT1 gene. This gene provides instructions for making a protein that is involved in adding sugar molecules to a protein called alpha dystroglycan. Alpha dystroglycan is important for stabilizing the muscle cell during contraction and relaxation. This protein is also found in the brain and spinal cord (central nervous system), eye, and other parts of the body.
The lack of sufficient Alpha dystroglycan protein, lead to a general weakness in all my body muscles, and sever weakness in the eye nerves causing adverse short vision (-8) and delay in my mental development.
I am so grateful and satisfied with the results and the big improvements I achieved in the last few years. Nothing makes me proud more than seeing the facial reaction of people that knew me in the beginning, and they can’t believe it now, seeing me setting balanced, able to see them, and talking to them in English and Arabic!!!
Although walking and lifting things is still a challenge, I promise you, I will never stop fighting and will keep adding new reasons to surprise more people!
❞

❝ قصتي لا تختلف كثيرًا عن باقي القصص خاصةً في مجتمعنا الشرقي، فبعد إنجاب 3 بنات و4 إجهاضات متكررة، رزقني الله بطفلين هارون وأحمد.عندما وصل عمر هارون 3 سنوات ونصف بدأت عنده أعراض المرض وكان ذلك في عام 2013. وقتها أحمد لم يكمل السنة بعد، وبدأنا رحلة التشخيص التي استمرت عاماً كاملاً لمعرفة المرض وتشخيصه.
وبعد هذه السنة عرفنا المرض الذي يعاني منه ابني، وبدأنا فوراً خطة العلاج، وقمنا بفحص أخيه أحمد أيضا، وكان الخبر لنا صاعقاً وصادماً: ابني أحمد يعاني من نفس المرض الذي يعاني منه أخوه هارون.
حاولت جاهدة أن أبحث عن العلاج خارج سلطنة عُمان، حتى سمعت عن توفر علاج تجريبي في الولايات المتحدة الأميركية، فقمت بمراسلتهم فوراً كي يخضعوا احمد للعلاج التجريبي، ولكن كان الرد منهم أنها مسألة وقت للحصول على الموافقة لبدء خطة العلاج، وفعلا تمت الموافقة على استخدام الدواء ولكن للأسف الشديد فقد كان سعر العلاج باهظ التكلفة ولا أستطيع حتى شراءه. وهذا أصابنا بالإحباط الشديد.
في أواخر عام 2017 بدأ المرض عند ابني أحمد وفي عام 2018 سافرت إلى ألمانيا للعلاج، ولكن للأسف مرض أحمد وصل مرحلة متقدمة من الأعراض التراجعية، وبعد أن جلست مع المختصين وجدت أن الدواء الذي يتوفر لديهم لا يعالج المرض كلياً وإنما هو إبطاء للأعراض المرضية التي يعاني منها أحمد ولو كان الدواء تم تقديمه في عمر مبكر من ولادته قبل أن يتقدم لديه المرض فإن العلاج سيكون أنفع والاستجابة ستكون أفضل.
عُدت بعدها إلى عُمان وأنا كلي رضى بما وهبني الله من نعمة أحتويها في بيتي وشفيعاً لي في آخرتي، فالحمد لله على كل حال وقصتي لم تنتهي، ولكن كانت بداية لي.
فتجربتي مع مرض أولادي حولتها من تجربة أعاني منها ومن الألم الذي أعيشه دوماً إلى نقلة نوعية في عالم الأمراض النادرة في سلطنة عمان.
❞

❝ اتحدث عن مرض ابني نجيب وعلى التحديات التي تواجهه
ولدي نجيب من مواليد ٢٠١٥، معه مرض نادر مرض ضمور العضلات دوشين، وعنده نقص في الغده الدرقية. يواجه صعوبة في المشي وانتفاخ وضعف في العضلات، يستخدم الادوية مثل الكورتيزون والثيروكسين والفيتامين وأدوية الكالسيوم وأدوية الحموضة. لديه حول في العينين وقمنا بعمل عملية الحول ونجحت العملية.
لا يوجد اي علاجات حالياً في سلطنة عمان فقط خارج البلاد كعلاج العقار الجيني.
نكتفي علاجه حالياً بالتأهيل والعلاج الطبيعي والوظيفي والنفسي والحسي، والتربية الخاصة، والنطق، والتخاطب.
نجيب يستطيع المشي والركض واللعب ويعتمد على نفسه في بعض وظائفه في البيت ويستطيع التحدث لكن لا يستطيع المشي واللعب والركض لفترات طويلة يشعر بالتعب وضعف العضلات والخمول..
❞

❝ Initially, everything was fine. My little girl was just like any other child, completely normal. She laughed, played, and brought immense joy to our family. However, over time, around the age of three, we noticed that she still wasn't speaking correctly. And from that moment on, we realized that she was gradually losing her ability to walk and maintain balance. Today, I will tell you our story, the story of my brave daughter who is affected by CLN6.
Challenges Faced:
Among the difficulties we encountered, epilepsy was one of the most challenging aspects. I had never witnessed seizures before and didn't know how to react. Additionally, my daughter experienced frequent insomnia, which made it difficult for her to get enough rest. She also had ongoing stomach issues that caused constipation. The panic attacks she experienced were also challenging to handle.
Learning to Adapt:
Despite these difficulties, I never gave up. I learned to manage these situations and establish a stable routine for my daughter. With time and experience, I gained a better understanding of her condition and her specific needs. I sought advice from healthcare professionals, conducted extensive research, and found support from other parents in similar situations. Through all of this, I discovered strategies to mitigate the effects of epilepsy, alleviate insomnia, and find solutions for her stomach problems.
Strength and Resilience:
Above all, our family remained strong and resilient in the face of adversity. We faced each obstacle with determination and love for our daughter. We didn't let the difficulties define us; instead, we armed ourselves with knowledge and sought innovative solutions to improve her quality of life. We created a loving environment where our daughter could thrive, surrounded by unconditional support and understanding.
Conclusion:
Although our journey hasn't been easy, our love, dedication, and perseverance have made a significant difference. We have shown that with patience, knowledge, and adaptability, it is possible to navigate the complexities of CLN6. We embraced our daughter's unique needs and encouraged her to overcome the obstacles that come her way. Together, we continue to grow stronger, cherishing every moment and celebrating our daughter's incredible strength.
❞

❝ كانت فرحتنا بقدوم طفلتي الصغيرة ليان لا توصف. كانت طفلة جميلة وبصحة جيدة.
عدنا بها الى المنزل بعد ان طمئنونا انها بصحة جيدة، ولكن قلبي لاحظ عليها بعض الاختلاف. كنت دائما اقارن نموها بإخوتها بكل مراحل عمرها، والاحظ عليها التأخر بالنمو والحركة. كانت قبل ان تتم ال٣ أشهر لا تتبع ولا ترى جيدا، وليطمئن قلبي الذي يملئه الشك قمت بأخذها لاستشاري عيون وهنا كانت أولى الصدمات. طفلتي التي لم تكمل ال٣ أشهر من العمر مصابة بالماء الابيض بعينيها وتحتاج لإجراء عملية بأسرع وقت ممكن، والحمدلله قمنا بإجراء العملية بعمر ال٣ أشهر.
.
وبعدها بدأ الشك يتغلغل في قلبي وكنت ألاحظ تأخرها بشكل كبير، كان عمرها ٩ أشهر ولا تستطيع القيام سوى التقلب على جانبيها وكانت نوبات ومشاكل الصدر تهاجمها بشكل متكرر. قمنا بزيارة عدة اطباء، ولكنهم كلهم كانوا يخبروني بأن الاطفال مختلفين بتطورهم وبأن ما اشعر به ما هو الا وساوس وان طفلتي صحية، ولكن قلبي كان يخبرني ان طفلتي تعاني من شيء الى ان طلب أحد الاطباء ان يتم نقلنا الى مستشفى مختص بالرياض وقمنا بأول فحص للجينات.
اخذ منا لاكتشاف ما تعانيه طفلتي من مرض قرابة السنة والنصف و٣ فحوصات جينيه، وعندما تم اكتشاف الجين المسبب للمرض جاءنا الاتصال كحبل النجاة الذي كنت احلم بالوصول اليه او هذا ما ظننته.
دخلنا لغرفة الدكتورة وبدأت تتمتم امامنا بعبارات لم أكن افهم منها شيء: جينات، مصاب، حامل للجين، متنحي ... كانت كلماتها غير مفهومة، لم افهم منها ما هو مرض طفلتي. اعطتني اسم المرض بورقة مكتوبة بخط يدها واخبرتني وهي تعطيني اياها ((إن الاطفال المصابين بهذا المرض لا يعمرون ويموتون بسن مبكرة، لا تعلقوا قلوبكم بها)). بهذه اللحظة شعرت كما لو ان حبل النجاة قد قطع، دخلت بحالة اكتئاب طويلة، ولكني تمكنت من الخروج منها عندما قررت ان ابحث عن المرض فأخذت الورقة المكتوبة بخط الدكتورة وكتبت اسم المرض وبدأت البحث، وتفاجأت بأنه لا توجد اي معلومات عنه باللغة العربية فقمت بتحسين لغتي الإنجليزية و بدأت ابحث بالمواقع الأجنبية وبمواقع التواصل الاجتماعي. وجدت العديد من المقالات ومجموعات الدعم والاسر التي تشارك قصصها. تمكنت من الوصول للعديد من الاسر وحصلت على العديد من المعلومات وتمكنت من ترجمة أول مقالة طبية عن مرض طفلتي للغة العربية ووضعتها بمدونة وراثة. بدأت بالبحث عن اسر لمصابين من نفس المرض بالوطن العربي وقمت بتجميعهم وتبادل المعلومات والتجارب والدعم النفسي معهم، تمكنت من التواصل مع الدكتورة التي اكتشفت الجين المسبب لمتلازمة طفلتي د. نانسي فردرمان وشاركت اون لاين بالمؤتمر السنوي لمتلازمة طفلتي rhizo conference.
كنت كلما اتعمق بفهم مرض طفلتي، كلما أوقن ان الله هو القادر على كل شيء وان كل ما بيدي هو ان أقدم لها الافضل من علاج وتأهيل واترك الباقي لله عز وجل، هو ألطف وأرحم بها وهو القادر على كل شيء.
طفلتي كانت ملهمتي ولازالت، بسببها تمكنت من التوعية بالأمراض النادرة بكل ما اوتيت من قوة وتمكنت من إقامة العديد من الفعاليات مع عدة جهات للتوعية بالأمراض النادرة وتمكنت من الوصول للعديد من الامهات والاهالي وتوعيتهم وتوجيههم ومساعدتهم على تخطي الصدمة والوقوف على أقدامهم والسعي لأطفالهم. قد أكون مجرد أم لطفلة بمرض نادر، ولكني أرى نفسي كأم لطفلة تحدت المستحيل ووقفت بابتسامتها الجميلة لتكمل حياتها بكل قوة وإصرار، طفلتي ليان هي ملهمتي.
❞

❝ Hello All
I am Zein, 14 years old. I have biochemically diagnosed with Mitochondrial Cytopathy affecting Complex V of respiratory chain enzymes along with epilepsy and development delay.
I discovered the disease when I was two years old, then gradually I lost all powers in my muscles and now I am in a wheelchair because the disease worked on weakening my muscles.
My family did two tests in the Netherlands, I sent my fibroblast, and the lab couldn't confirm the disease. Doctors told my family that they can confirm only the gene and to deal with the clinical symptoms.
I lost all my skills I once had, I have difficulties with speech, I have weak muscles especially upper area so I can't feed and dress myself, hold a pen, or brush my hair.
I have scoliosis and another main problem is focal epilepsy.
My family supports me so much. Intellectually I am a bright boy who attend schooling primarily from home under my mother supervision which will help me with improving my quality of life. I participated in many initiatives, and I created my own social media account to show the community what I am able to do and say.
Unfortunately, there is no treatment for most mitochondrial diseases, however; hope exists with scientific achievements and the diversity of research and medicine.
❞
MENA REGION

MENA Organization for Rare Diseases was established to serve and support people with rare diseases in the region of Middle East and North Africa (MENA).
People with rare diseases and their families face significant challenges that arise from the infrequency of their medical conditions, such as limited knowledge about their diseases, long diagnostic journey, and inadequate clinical management and support available for them.
A disease is considered rare if it has a prevalence of less than 1 in 2000. Recent studies indicate that there are more than 10,000 different rare diseases. Although they are uncommon individually, collectively they affect more than 5% of the population. Unfortunately, only 5% of rare diseases have available therapies.
The Middle East and North African (MENA) region includes more than 20 countries with an area of 15 million square kilometers and more than 600 million population. This region shares cultural factors causing higher prevalence of rare diseases such the common practices of large family size, high maternal and paternal age, and high consanguinity rates.
MENA Organization for Rare Diseases was established in Dubai to support and advocate for rare diseases in the region of Middle East and North Africa (MENA). The goals of MENA Organization for Rare Diseases are to educate, connect, and support.
EDUCATE
MENA Organization for Rare Diseases aims to educate, spread knowledge, and increase awareness about rare diseases among healthcare providers, patients and their families, and the public. This goal can be achieved through scientific conferences, family meetings, workshops, training programs, social media posts, and online tools such as electronic newsletters and educational webpages and videos.
CONNECT
MENA Organization for Rare Diseases aims to connect people to exchange knowledge and experience about rare diseases. This can be achieved by organizing social events and establishing rare disease databases and support groups to connect patients and families with rare diseases. We also establish databases and networks for healthcare professionals providing care for patients with rare diseases.
SUPPORT
MENA Organization for Rare Diseases aims to provide support for individuals with rare diseases and their families through connecting patients to healthcare providers and facilitating receiving services from community services and healthcare institutions such as hospitals, clinics, laboratories, and rehabilitation centers.